Las miopatías mitocondriales comprenden un grupo heterogéneo de trastornos primarios donde la disfunción mitocondrial altera de forma directa y considerable la bioenergética celular. Estas enfermedades se distinguen porque la alteración de la función de las mitocondrias basta para explicar la clínica y la fisiopatología, afectando principalmente el músculo esquelético y, en muchos casos, diversos órganos y sistemas. Analizar los elementos estructurales y funcionales implicados permite descomponer los mecanismos celulares y moleculares que subyacen a su presentación clínica variable.
🧠 Idea central
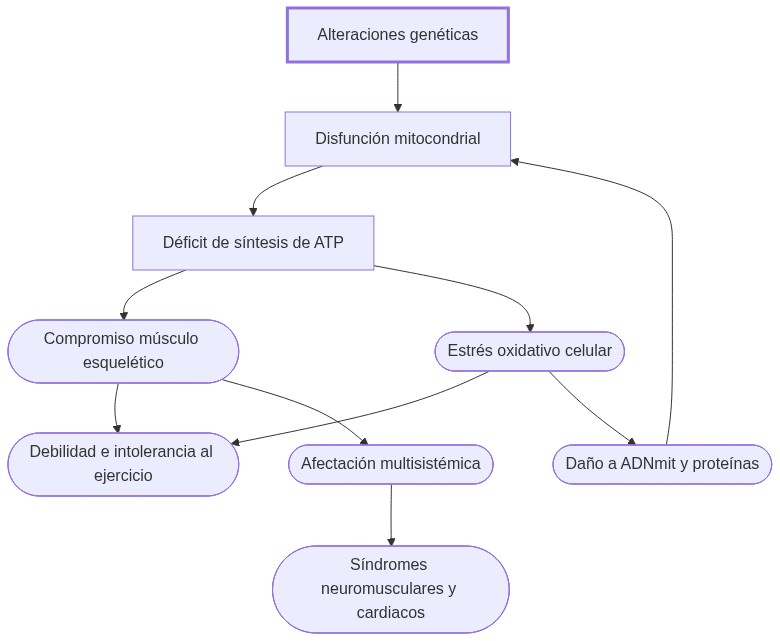
Las miopatías mitocondriales surgen a partir de disfunciones que comprometen la función esencial de las mitocondrias: generar adenosín trifosfato (ATP) mediante fosforilación oxidativa. Este déficit energético impacta con mayor severidad a los tejidos con alta demanda metabólica, como músculo esquelético, sistema nervioso central y corazón. Además de alteraciones moleculares y bioquímicas, influyen mutaciones en genes mitocondriales (ADNmt) o nucleares, contribuyendo a una gran variabilidad fenotípica y evolutiva. La fisiopatología radica en la relación entre las alteraciones estructurales del orgánulo y el déficit funcional, que se manifiesta desde intolerancia al ejercicio hasta síndromes multisistémicos graves.
🌍 Contexto y alcance
El análisis de las miopatías mitocondriales abarca desde el nivel subcelular hasta manifestaciones en órganos y sistemas. El contexto celular incluye mutaciones tanto en el material genético mitocondrial como nuclear, responsables de codificar proteínas involucradas en la cadena de transporte electrónico, el ensamblaje de complejos enzimáticos y la replicación del genoma mitocondrial.
Estas alteraciones pueden causar enfermedades que se expresan casi exclusivamente en músculo esquelético (“miopatías puras”), síndromes que afectan músculo y sistema nervioso (“encefalomiopatías mitocondriales”) o múltiples órganos. La enfermedad puede ser la manifestación principal o parte de cuadros sistémicos, con un rango clínico que va desde debilidad leve hasta fallo multiorgánico.
Epidemiológicamente, las miopatías mitocondriales pueden ser más frecuentes de lo previamente considerado, especialmente cuando la afectación muscular es predominante o acompañante.
🧬 Estructuras clave
Para comprender cómo las alteraciones moleculares generan enfermedad, es clave identificar las siguientes estructuras y su función relevante:
- Mitocondria: Orgánulo subcelular responsable de la generación de energía. En el músculo, su abundancia se relaciona con alta demanda metabólica.
- Membrana interna mitocondrial: Contiene la cadena respiratoria y establece el gradiente electroquímico necesario para la síntesis de ATP.
- Complejos enzimáticos I-IV y ATP sintasa: Proteínas que catalizan la transferencia de electrones y acoplan la síntesis de ATP mediante fosforilación oxidativa.
- ADN mitocondrial (ADNmt): Genoma circular pequeño que codifica proteínas esenciales de la cadena respiratoria y ARN de transferencia y ribosomal.
- ADN nuclear: Codifica la mayoría de las proteínas mitocondriales, incluidas las implicadas en ensamblaje, replicación y dinámica mitocondrial.
- Matriz mitocondrial: Espacio interno donde ocurren rutas metabólicas como ciclo de Krebs y β-oxidación de ácidos grasos.
Estas entidades se interrelacionan para mantener la homeostasis energética y el metabolismo intermediario en las células musculares y otros tejidos afectados.
| Estructura | Qué es | Dónde actúa | Función relevante |
|---|---|---|---|
| Membrana interna | Bicapa lipídica con proteínas integrales | Mitocondrias de todas las células nucleadas | Aloja la cadena respiratoria y mantiene el gradiente de protones |
| Complejos I-IV | Agregados multiproteicos | Membrana interna mitocondrial | Transfieren electrones sucesivamente y bombean protones |
| ATP sintasa | Enzima transmembrana (Complejo V) | Membrana interna mitocondrial | Sintetiza ATP mediante gradiente de protones |
| ADN mitocondrial | Molécula de ADN circular | Matriz mitocondrial | Códigos para 13 proteínas, 22 tRNA, 2 rRNA esenciales para función mitocondrial |
| Matriz mitocondrial | Espacio interno viscoso | Centro de la mitocondria | Sitio de ciclo de Krebs y otras rutas metabólicas |
⚙️ Funciones y procesos
La fisiología mitocondrial que sustenta la función muscular normal implica varios procesos interconectados:
-
Producción de ATP (adenosín trifosfato):
- En la matriz mitocondrial, rutas como el ciclo de Krebs y la β-oxidación generan NADH y FADH2.
- Estos intermediarios entregan electrones al Complejo I (NADH) o II (FADH2) y luego a la ubiquinona (Coenzima Q).
- Los electrones se transfieren por los Complejos III y IV formando un flujo que permite el bombeo de protones al espacio intermembrana.
- El bombeo de protones (H+) genera un gradiente electroquímico (fuerza protón-motriz) a través de la membrana interna.
- La ATP sintasa (Complejo V) utiliza este gradiente para sintetizar ATP a partir de ADP y fosfato inorgánico.
-
Expresión genética dual:
- La mayoría de las proteínas mitocondriales se sintetizan en el citoplasma a partir de genes nucleares e ingresan postraducción a la mitocondria.
- El ADNmt codifica solo 13 proteínas, todas componentes de la cadena respiratoria; su alteración afecta la eficiencia de los complejos.
-
Determinantes del flujo energético:
- La cantidad y distribución de mitocondrias dependen del tipo de fibra muscular (oxidativa o glucolítica).
- El flujo energético puede verse afectado por defectos que limiten la transferencia electrónica, el mantenimiento del gradiente o síntesis de ATP.
-
Homeostasis redox y especies reactivas de oxígeno:
- Cuando la cadena respiratoria se enlentece, pueden generarse especies reactivas de oxígeno que producen estrés oxidativo.
- La acumulación sostenida de estas especies daña lípidos, proteínas y ADNmt, empeorando la disfunción mitocondrial.
Mutaciones que afectan la síntesis o función de complejos enzimáticos comprometen la fosforilación oxidativa, disminuyen la producción de ATP y generan crisis energéticas en tejidos vulnerables.
🔗 Integración funcional
La fisiopatología de las miopatías mitocondriales se entiende integrando los siguientes aspectos:
- Mutaciones en ADNmt o nuclear: Alteran la síntesis o ensamblaje correcto de complejos respiratorios, reduciendo la eficiencia del transporte de electrones y la capacidad de mantener el gradiente de protones, disminuyendo crónicamente la producción de ATP.
- Fallo en la bomba de protones: La disminución del gradiente afecta la actividad de la ATP sintasa, ralentizando la síntesis de ATP y comprometiendo funciones energéticas como la contracción muscular.
- Acumulación de productos intermedios: El flujo inadecuado de electrones favorece la generación de radicales libres que dañan las células musculares y otros órganos.
- Compromiso en ensamblaje estructural: Genes nucleares regulan replicación y reparación del ADNmt y ensamblaje de proteínas mitocondriales; su alteración genera déficit cuantitativo (disminución de mitocondrias funcionales) y cualitativo (alteraciones estructurales).
- Heteroplasmia: La coexistencia de mitocondrias normales y mutadas determina el fenotipo clínico según la proporción mutacional en cada tejido; los órganos presentan variabilidad en la afectación.
- Afección de tejidos no musculares: El corazón, sistema nervioso central y órganos endocrinos también pueden afectarse, manifestando disfunciones específicas como arritmias o encefalopatías.
La alteración de estos procesos produce en el músculo esquelético debilidad variable, intolerancia al ejercicio y manifestaciones clínicas que pueden ir de asintomáticas a graves, especialmente bajo estrés fisiológico o metabólico.
🔬 Métodos y evidencias
El diagnóstico de una miopatía mitocondrial requiere una evaluación integral que considere aspectos clínicos, bioquímicos y moleculares:
- Evaluación clínica y antecedentes familiares: Historia multisistémica y patrones de herencia (materna, autosómica) sugieren origen mitocondrial. Importa documentar síntomas musculares y signos en otros sistemas.
- Estudios de laboratorio: Medición de lactato y piruvato (que pueden elevarse), creatina quinasa (normal o elevada), y otros marcadores metabólicos.
- Biopsia muscular: Permite observar fibras musculares con acumulación anómala de mitocondrias (“fibras rojas desgarradas” en tinción de Gomori tricrómico) y fibras negativas para citocromo c oxidasa, indicando alteración mitocondrial.
- Estudios histoquímicos: Tinciones para NADH, succinato deshidrogenasa (SDH), y citocromo c oxidasa (COX) indican proliferación mitocondrial y alteraciones específicas.
- Análisis genético: Identifica variantes patogénicas en ADNmt o nuclear; la heteroplasmia y depleción de ADNmt se evalúan en tejidos según el caso, mediante secuenciación de nueva generación (NGS).
- Análisis bioquímico: Medición de actividad en complejos respiratorios en tejidos frescos, útil cuando el análisis genético es inconcluso, aunque con limitaciones técnicas.
- Pruebas funcionales: Ejercicio controlado con medición de lactato y parámetros metabólicos puede ser una pista indirecta para déficit mitocondrial muscular en pacientes con intolerancia al ejercicio.
La integración de hallazgos estructurales, bioquímicos y genéticos permite confirmar la naturaleza mitocondrial del déficit muscular.
🩺 Puente clínico
El conocimiento de la biología mitocondrial facilita la relación entre mecanismos moleculares y manifestaciones clínicas:
- Déficit de ATP: La insuficiente producción energética genera debilidad muscular progresiva, intolerancia al ejercicio y, en casos avanzados, rabdomiólisis.
- Heteroplasmia y variabilidad clínica: La expresión clínica depende de la proporción de mitocondrias normales y mutadas en cada tejido.
-
Categorías clínicas:
- Miopatía pura aislada: Limitada a músculo esquelético (intolerancia al ejercicio, debilidad proximal, mialgias).
- Oftalmoplejia externa progresiva crónica y síndrome de Kearns-Sayre: Paresia progresiva de músculos extraoculares y ptosis, con posibles manifestaciones multisistémicas en Kearns-Sayre.
- Miopatía en síndromes multisistémicos: Ejemplos incluyen MELAS, MERRF, Leigh, MNGIE, entre otros.
- Factores moduladores: Estrés fisiológico (infección, ayuno, cirugía) puede precipitar o agravar síntomas. Signos musculares pueden coexistir con neuropatía, ataxia, pérdida auditiva, alteración cardíaca o endocrina, según síndrome genético.
Reconocer estas relaciones es fundamental para interpretar las manifestaciones clínicas en enfermedades donde la fisiopatología mitocondrial es central.
💎 Perlas de alto rendimiento
- Las mitocondrias generan ATP mediante fosforilación oxidativa, proceso esencial en músculo esquelético por su alta demanda energética.
- Las mutaciones en ADN mitocondrial o nuclear afectan la bioenergética celular y pueden causar miopatías mitocondriales.
- El compromiso de complejos respiratorios disminuye la síntesis de ATP y favorece la producción de especies reactivas de oxígeno.
- La heteroplasmia explica la variabilidad fenotípica según la proporción de mitocondrias mutadas en cada tejido.
- Los hallazgos histológicos clave incluyen fibras rojas desgarradas y fibras COX-negativas en biopsias musculares.
- El diagnóstico molecular requiere a menudo muestras de tejido afectado debido a la variabilidad en la presencia de mutaciones en sangre.
- Las mitocondrias poseen ADN propio, pero la mayoría de proteínas funcionales derivan de genes nucleares.
- Los síndromes clásicos incluyen MELAS, MERRF, Leigh, MNGIE, y Kearns-Sayre, con variadas manifestaciones musculares y multisistémicas.
🧠 Puntos clave
- Las miopatías mitocondriales resultan de defectos genéticos que alteran la fosforilación oxidativa y la síntesis de ATP en músculo y otros tejidos.
- Las mutaciones pueden afectar ADNmt o nuclear, comprometiendo producción, ensamblaje y función de complejos respiratorios.
- El déficit energético afecta principalmente tejidos con alta demanda metabólica como músculo, sistema nervioso y corazón.
- La heteroplasmia y la proporción de mitocondrias normales y mutadas determinan la expresión clínica y severidad de la enfermedad.
- El diagnóstico integra criterios clínicos, bioquímicos, histológicos y genéticos para confirmar la disfunción mitocondrial.
- El estrés fisiológico puede revelar o intensificar manifestaciones subclínicas debido a la limitada reserva bioenergética.
❓ Preguntas frecuentes
-
¿Por qué las miopatías mitocondriales afectan preferentemente músculos y sistema nervioso central?
Porque estos tejidos tienen alta densidad mitocondrial y demanda energética constante, por lo que su función se compromete rápidamente ante déficit de ATP. -
¿Cómo influye la heteroplasmia en la expresión clínica?
La proporción de mitocondrias mutadas en cada célula afecta el umbral funcional; una mayor carga mutacional puede desencadenar síntomas mientras que una carga baja puede ser asintomática. -
¿Cuál es el origen de las fibras rojas desgarradas en biopsia muscular?
Son causadas por la proliferación compensatoria de mitocondrias anormales subsarcolemmales, visibles con la tinción de Gomori tricrómico. -
¿Por qué unas miopatías mitocondriales se presentan aisladas y otras multisistémicas?
Depende del gen y tipo de mutación, el nivel de heteroplasmia en cada tejido, y las necesidades metabólicas locales, lo que condiciona el grado y distribución del daño. -
¿Qué diferencias hay entre mutaciones en ADN mitocondrial y ADN nuclear?
Las mutaciones en ADNmt se heredan principalmente por línea materna y muestran heteroplasmia; las del ADN nuclear pueden seguir herencia autosómica dominante o recesiva y afectan funciones esenciales del ensamblaje y mantenimiento mitocondrial. -
¿Qué hallazgos bioquímicos sugieren una miopatía mitocondrial?
Elevación de lactato y piruvato en sangre o líquido cefalorraquídeo, acidosis metabólica, y creatina quinasa normal o levemente elevada, aunque estos hallazgos no son específicos. -
¿Cómo contribuye el estrés oxidativo al daño tisular?
La fuga de electrones genera radicales libres que dañan lípidos, proteínas y ADNmt, exacerbando la disfunción mitocondrial y el daño celular. -
¿Por qué a veces se requiere biopsia muscular para diagnóstico molecular?
Porque la heteroplasmia puede variar y algunas mutaciones no son detectables en sangre periférica, requiriendo tejido afectado para confirmar la alteración genética.
📚 Referencias
Evaluación Interactiva Progresiva
Este cuestionario evalúa la comprensión del contenido biológico relacionado con las miopatías mitocondriales presentado en el artículo.
Nivel 1 – Básico
¿Cuál es la función principal de la mitocondria en el músculo esquelético según el artículo?
Según la tabla, ¿qué función relevante tiene la membrana interna mitocondrial?
¿Qué estructuras codifica el ADN mitocondrial (ADNmt) según el artículo?
Nivel 2 – Intermedio
¿Cuál es la relación correcta entre ADN nuclear y núcleo de la disfunción mitocondrial?
¿Cuál de estas afirmaciones describe correctamente una característica de la heteroplasmia según el texto?
Según el artículo, ¿cuál es la diferencia fundamental entre las miopatías puras y las encefalomiopatías mitocondriales?
Nivel 3 – Avanzado
¿Qué consecuencia funcional genera una mutación que disminuye la eficiencia del bombeo de protones por los complejos respiratorios?
¿Cuál es el mecanismo detallado para la producción de ATP mediante fosforilación oxidativa en mitocondrias según el artículo?
¿Cómo afecta la acumulación de especies reactivas de oxígeno (ERO) al músculo en miopatías mitocondriales?
Contenido educativo. No sustituye la enseñanza formal ni el juicio clínico.